PUBLICATIONS
Dolutegravir restores gut microbiota in late-stage HIV-1 unlike darunavir: an open-label, randomized clinical trial
Authors: Francesc Català-Moll, Carlos Blázquez-Bondia, Judit Farré-Badia, Ferran Torres, Christian Manzardo, Eva Bonfill, Adrià Curran, Pere Domingo, Daniel Podzamczer, Lluís Force, Vicenç Falcó, Mariona Parera, Maria Casadellà, José Maria Miró, Marc Noguera-Julian, Roger Paredes & ADVANZ-4 MISTRAL investigators.
Abstract: Late presentation of HIV-1 infection is linked to gut dysbiosis, impaired immune reconstitution, excess inflammation, immune activation, and increased morbidity and mortality. It is unclear if antiretroviral therapy initiation can reverse HIV-associated gut dysbiosis at all, or if specific antiretroviral regimens are more effective in restoring the gut microbiota than others. This has important implications for the long-term health of individuals with HIV. In this multicenter, open-label, randomized clinical trial (NCT02337322), 88 antiretroviral-naïve individuals with advanced HIV-1 infection (median CD4+ T cells of 34 cells/mm3) were randomized (1:1) to initiate lamivudine/abacavir plus either dolutegravir or ritonavir-boosted darunavir, and were followed for 2 years. Both groups had similar HIV-1 suppression rates and recovery of CD4+ T cells. However, treatment with dolutegravir led to increased gut microbial richness and diversity and enrichment of specific microbial taxa and metabolic pathways. These changes were associated with reduced inflammation and lower immune activation, outcomes that did not occur with darunavir/ritonavir. After two years, participants on dolutegravir-based therapy had gut microbiota profiles more closely resembling those of people without HIV, compared to individuals taking darunavir/ritonavir. In summary, dolutegravir-based therapy restores the gut microbiota more effectively than darunavir/ritonavir in patients who present late with HIV.
The cervicovaginal microbiome associates with spatially restricted host transcriptional signatures throughout the human ectocervical epithelium and submucosa
Authors: Vilde Kaldhusdal, Mathias Franzén Boger, Adam D. Burgener, Julie Lajoie, Kenneth Omollo, Joshua Kimani, Annelie Tjernlund, Keith R. Fowke, Douglas S. Kwon, Gabriella Edfeldt, Kristina Broliden
Abstract:
The cervicovaginal microbiome is a key biological determinant of human immuno deficiency virus (HIV) susceptibility, but its underlying impact on the ectocervical transcriptional landscape is unclear. Ectocervical tissue samples from Kenyan female sex workers were categorized into pre-defined cervicovaginal microbiome groups based on dominant compositions: Lactobacillus crispatus/acidophilus, Lactobacillus iners, Gardnerella, and ‘highly diverse’. The tissue samples (n=21) were assessed using spatial transcriptomics, revealing three epithelial, one mixed border, and nine submucosal gene clusters. Differential gene expression analysis across the microbiome groups and gene clusters identified 3,771 unique genes. The highly diverse microbiome group associated with the largest differences, mostly located near the epithelial basal membrane, encompassing genes involved in epithelial maintenance, submucosal extracellular matrix structures, and immune function. The L. crispatus/ acidophilus-dominated group was identified by genes involved in active immune engagement, supporting mucosal barrier integrity. Weighted gene co-expression analysis confirmed tissue-wide altered gene expression associated with all microbiome groups and with individual bacterial taxa. Despite the assumption that microbiome colonization is restricted to the luminal surface, the transcriptional landscape was affected throughout the mucosa, with the most pronounced effect near both sides of the basal membrane. This broad association with the mucosal barrier integrity could affect susceptibility to HIV acquisition.
Immune reconstitution in very advanced HIV patients treated with dolutegravir vs. darunavir-based triple antiretroviral therapy: the Advanz-4 randomized clinical trial
Authors: Jose M. Miro, Ferran Torres, Christian Manzardo, Eva Bonfill, Adrià Curran, Pere Domingo, Daniel Podzamczer, Roger Paredes, Lluis Force, Vicenç Falco, Mar Gutierrez, Maria Saumoy, Anna Castelli, Alexy Inciarte, Cristina Rovira, Anna Cruceta, Carmen Hurtado, Núria Climent, Francisco Lozano, Montserrat Plana and the Advanz-4 investigators
Abstract: The aim of this trial was to compare the immune reconstitution, virologic response, and safety of dolutegravir (DTG) vs. darunavir (DRV) boosted-based antiretroviral (ART) regimens in very advanced HIV patients. Methods: Phase IV, randomized (1:1 ratio), open-label trial, including adult (≥18 years) ART-naïve HIV1+ patients with CD4+ cell counts <100 cells/μL from nine hospitals in Spain. Participants were
randomized to lamivudine (3TC)/abacavir (ABC)/DTG (DTG arm) and 3TC/ABC/DRV + ritonavir (DRV/r arm). Primary endpoint: change in the absolute CD4+ cell number at 48 weeks in the modified
intention-to-treat population. Trial Registration: NCT02337322.
Results: In total, 104 patients (86.5% male, median (interquartile range [IQR]) age 41.0 (31.5, 47.0) years) were recruited and randomized to DTG (n = 52) and DRV/r arms (n = 52). Baseline median (IQR) CD4+ cell counts were 39.5 (16.0, 72.9) and 29.0 (8.0, 60.0) cells/μL in the DTG and DRV/r arms. They significantly increased by median (IQR) 206.5 (154.4, 310.5) and 180.0 (89.4, 314.0) cells/μL, respectively (p 0.2549); 29 (55.8%) and 21 (42.9%) (p 0.2343) patients, respectively, reached >200 cells/μL. Of the patients, 41 (78.8%) and 31 (63.3%) (p 0.1229) in the DTG and DRV/r arms, respectively, achieve undetectable viral loads at 48 weeks; differences were significant at weeks 4 (p < 0.0001) and 12 (p 0.0009). Inflammation and bacterial translocation markers decreased more in the DTG arm, median (IQR) − 8 (− 11, − 4) vs. − 5 (− 9, − 3) pg/mL (p 0.0357) and − 972 (− 1334, − 508) vs. − 544 (− 1128, − 292) μg/mL (p 0.0565), respectively, at 48 weeks. Discontinuation rates were higher in the DRV/r arm (3/52 (5.8%) vs. 9/51 (18.4%); p 0.0526).
Conclusions: DTG/3TC/ABC is safe and efficacious in very advanced ART-naïve HIV + patients, induced a faster virologic response, and was superior to the DRV/r regimen in reducing inflammation and bacterial translocation markers at 48 weeks
Interactions between gut microbiota, plasma metabolome and brain function in the setting of a HIV cure trial
Authors: Alessandra Borgognone, Anna Prats, Ashish Arunkumar Sharma, Ignacio Martinez-Zalacaín, Carles Soriano-Mas, Christian Brander, Bonaventura Clotet, José Moltó, Beatriz Mothe, Rafick-Pierre Sekaly, Roger Paredes and Jose A. Muñoz-Moreno.
Abstract:
Introduction: The intestinal microbiota composition has been linked to neurocognitive impairment in people with HIV (PWH). However, the potential interplay of microbial species and related metabolites, particularly in the context of an HIV cure strategy remains underexplored. The BCN02 trial evaluated the impact of romidepsin (RMD), used as a HIV-1 latency reversing agent and with reported beneficial neurological effects, combined with the MVA.HIVconsv vaccine on virus control during 32-weeks of monitored antiretroviral treatment interruption (MAP) in early-treated HIV-infected individuals. Here, we analyzed longitudinal gut microbiome, plasma metabolome and brain functioning data to identify potential associations and novel putative biomarkers of HIV-associated neurocognitive disorders (HAND).
Methods: Data from fecal shotgun metagenomics, plasma metabolome, cognitive (standardized neuropsychological test score covering 6 cognitive domains, NPZ-6), functional (neuropsychiatric symptoms) and neuroimaging assessments were obtained and evaluated in 18 participants before and after RMD administration, and at the study end (post-MAP follow-up) in the BCN02 trial.
Results: Participants with neurocognitive impairment (Lower vs. Higher NPZ-6 score group) were enriched in bacterial species, including Desulfovibrio desulfuricans, Sutterella wadsworthensis and Streptococcus thermophilus, and showed higher 1,2-propanediol degradation microbial pathway levels, before RMD administration. A multi-omics profiling showed significant and positive correlations between these microbial features and lipid-related metabolic pathways, previously linked to neurological disorders (i.e., sphingolipid, ether lipid, and glycerophospholipid metabolism), in participants with neurocognitive impairment, before RMD administration. Three indices (microbial, metabolite based and combined) obtained from the discriminant features were assessed longitudinally, showing progressive similarities between NPZ-6 score groups over time. Furthermore, the three indices and related discriminant features correlated negatively with functional outcomes, such as quality of life and daily functioning, and positively with depression, stress and CNS-related symptoms before RMD administration, while these associations became less discernible at the subsequent timepoints.
Conclusion: While the direct effect of the intervention on the observed shifts cannot be conclusively determined in this study settings, these findings strengthen the link between gut bacteria, related metabolites, and neurocognitive function in PWH, and provide an analytical framework for future validation studies aimed at discovering predictive biomarkers for neurocognitive impairment in PWH.
DOI: https://doi.org/10.3389/fcimb.2025.1629901
JOURNAL: Frontiers in Cellular and Infection Immunology
DATE: 2025 Aug 20
Spatial transcriptomics and in situ immune cell profiling of the host ectocervical landscape of HIV infected Kenyan sex working women
Authors: Mathias Franzén Boger, Vilde Kaldhusdal, Anna Pascual-Reguant, Sandy Kroh, Ralf Uecker, Adam D.Burgener, Julie Lajoie, Kenneth Omollo, Joshua Kimani, Keith R.Fowke, Anja E.Hauser, Annelie Tjernlund, Kristina Broliden
Abstract:
Introduction: Chronic immune activation is a hallmark of human immunodeficiency virus (HIV) infection that significantly impacts disease pathogenesis. However, in-depth studies characterizing the immunological landscape of the ectocervix during chronic HIV infection remain scarce despite the importance of this tissue site for HIV transmission.
Methods: Ectocervical tissue samples were obtained from antiretroviral-naïve HIV-seropositive and -seronegative Kenyan female sex workers. These samples were assessed by spatial transcriptomics and Gene Set Enrichment Analysis. We further performed multi-epitope ligand cartography (MELC) using an in situ staining panel that included 17 markers of primarily T cell–mediated immune responses.
Results: Spatial transcriptomics revealed tissue-wide immune activation encompassing immune responses associated with chronic HIV infection. First, both the epithelial and submucosal compartments showed diverse but significant upregulation of humoral immune responses, as indicated by the expression of several antibody-related genes. Second, an antiviral state–associated cellular immunity was also observed in the HIV-seropositive group, characterized by upregulation of genes involved in interferon signaling across the mucosal tissue and a more spatially restricted mucosal expression of genes related to T cell activity and effector functions relative to the HIV-seronegative group. Additionally, HIV associated structural alterations were evident within both compartments. Downregulated genes across the epithelium were mainly linked to epithelial integrity, with the outer layer involved in terminal differentiation and the inner layer associated with epithelial structure. MELC analysis further revealed a significantly increased ectocervical leukocyte population in HIV-seropositive participants, primarily driven by an increase in CD8+ T cells while the CD4+ T cell population remained stable. Consistent with our spatial transcriptomics data, T cells from HIV-seropositive participants showed an increased effector phenotype, defined by elevated expression of various granzymes.
Conclusion: By combining spatial transcriptomics and MELC, we identified significant HIV-associated cervical immune activity driven by induction of both T and B cell activity, together with a general antiviral state characterized by sustained interferon induction. These findings underscore that chronic HIV infection is associated with an altered ectocervical mucosal immune landscape years after primary infection. This sheds light on HIV pathogenesis at distant local sites and complements current knowledge on HIV-associated systemic immune activation.
Estradiol-mediated enhancement of the human ectocervical epithelial barrier correlates with desmoglein-1 expression in the follicular menstrual phase
Authors: Frideborg Bradley, Alexandra Stern, Mathias Franzén Boger, Zaynab Mousavian, Olga Dethlefsen, Vilde Kaldhusdal, Julie Lajoie, Kenneth Omollo, Sofia Bergström, Anna Månberg, Peter Nilsson, Joshua Kimani, Adam D. Burgener, Annelie Tjernlund, Christopher Sundling, Keith R. Fowke and Kristina Broliden
ABSTRACT:
Background
The cervicovaginal epithelial barrier is crucial for defending the female reproductive tract against sexually transmitted infections. Hormones, specifically estradiol and progesterone, along with their respective receptor expressions, play an important role in modulating this barrier. However, the influence of estradiol and progesterone on gene and protein expression in the ectocervical mucosa of naturally cycling women is not well understood.
Methods
Mucosal and blood samples were collected from Kenyan female sex workers at high risk of sexually transmitted infections. All samples were obtained at two time points, separated by two weeks, aiming for the follicular and luteal phases of the menstrual cycle. Ectocervical tissue biopsies were analyzed by RNA-sequencing and in situ immunofluorescence staining, cervicovaginal lavage samples (CVL) were evaluated using protein profiling, and plasma samples were analyzed for hormone levels.
Results
Unsupervised clustering of RNA-sequencing data was performed using Weighted gene co-expression network analysis (WGCNA). In the follicular phase, estradiol levels positively correlated with a gene module representing epithelial structure and function, and negatively correlated with a gene module representing cell cycle regulation. These correlations were confirmed using regression analysis including adjustment for bacterial vaginosis status. Using WGCNA, no gene module correlated with progesterone levels in the follicular phase. In the luteal phase, no gene module correlated with either estradiol or progesterone levels. Protein profiling on CVL revealed that higher levels of estradiol during the follicular phase correlated with increased expression of epithelial barrier integrity markers, including DSG1. This contrasted to the limited correlations of protein expression with estradiol levels in the luteal phase. In situ imaging analysis confirmed that higher estradiol levels during the follicular phase correlated with increased DSG1 expression.
Conclusions
We demonstrate that estradiol levels positively correlate with specific markers of ectocervical epithelial structure and function, particularly DSG1, during the follicular phase of the menstrual cycle. Neither progesterone levels during the follicular phase nor estradiol and progesterone levels during the luteal phase correlated with any specific sets of gene markers. These findings align with the expression of estradiol and progesterone receptors in the ectocervical epithelium during these menstrual phases.
Gut resistome linked to sexual preference and HIV infection
Authors: Elisa Rubio-Garcia, Maria Casadellà, Mariona Parera, Jordi Vila, Roger Paredes, Marc Noguera-Julian
ABSTRACT:
Background
People living with HIV (PLWH) are at increased risk of acquisition of multidrug resistant organisms due to higher rates of predisposing factors. The gut microbiome is the main reservoir of the collection of antimicrobial resistance determinants known as the gut resistome. In PLWH, changes in gut microbiome have been linked to immune activation and HIV-1 associated complications. Specifically, gut dysbiosis defined by low microbial gene richness has been linked to low Nadir CD4 + T-cell counts. Additionally, sexual preference has been shown to strongly influence gut microbiome composition in PLWH resulting in different Prevotella or Bacteroides enriched enterotypes, in MSM (men-who-have–sex-with-men) or no-MSM, respectively. To date, little is known about gut resistome composition in PLWH due to the scarcity of studies using shotgun metagenomics. The present study aimed to detect associations between different microbiome features linked to HIV-1 infection and gut resistome composition.
Results
Using shotgun metagenomics we characterized the gut resistome composition of 129 HIV-1 infected subjects showing different HIV clinical profiles and 27 HIV-1 negative controls from a cross-sectional observational study conducted in Barcelona, Spain. Most no-MSM showed a Bacteroides-enriched enterotype and low microbial gene richness microbiomes. We did not identify differences in resistome diversity and composition according to HIV-1 infection or immune status. However, gut resistome was more diverse in MSM group, Prevotella-enriched enterotype and gut micorbiomes with high microbial gene richness compared to no-MSM group, Bacteroides-enriched enterotype and gut microbiomes with low microbial gene richness. Additionally, gut resistome beta-diversity was different according to the defined groups and we identified a set of differentially abundant antimicrobial resistance determinants based on the established categories.
Conclusions
Our findings reveal a significant correlation between gut resistome composition and various host variables commonly associated with gut microbiome, including microbiome enterotype, microbial gene richness, and sexual preference. These host variables have been previously linked to immune activation and lower Nadir CD4 + T-Cell counts, which are prognostic factors of HIV-related comorbidities. This study provides new insights into the relationship between antibiotic resistance and clinical characteristics of PLWH.
Impact of ChAdOx1 or DNA Prime Vaccination on Magnitude, Breadth, and Focus of MVA-Boosted Immunogen-Specific T Cell Responses
Authors: Olvera, A.; Romero-Martin, L.; Oriol-Tordera, B.; Rosas-Umbert, M.; Escribà, T.; Mothe, B.; Brander, C.
Abstract
The efficacy of anti-viral T-cell vaccines may greatly depend on their ability to generate high-magnitude responses targeting a broad range of different epitopes. Recently, we created the HIV T-cell immunogen HTI, designed to generate T-cell responses to protein fragments more frequently targeted by HIV controllers. In the present study, we aim to maximize the breadth and magnitude of the T-cell responses generated by HTI by combining different vaccine vectors expressing HTI. We evaluated the ability to induce strong and broad T-cell responses to the HTI immunogen through prime vaccination with DNA plasmid (D) or Chimpanzee Adenovirus Ox1 (ChAdOx1; C) vectors, followed by a Modified Virus Ankara (MVA; M) vaccine boost (DDD, DDDM, C, and CM). HTI-specific T-cell responses after vaccination were measured by IFN-γ-ELISpot assays in two inbred mice strains (C57BL/6 and BALB/c). CM was the schedule triggering the highest magnitude of the response in both mice strains. However, this effect was not reflected in an increase in the breadth of the response but rather in an increase in the magnitude of the response to specific immunodominant epitopes. Immunodominance profiles in the two mouse strains were different, with a clear dominance of T-cell responses to a Pol-derived peptide pool after CM vaccination in C57BL/6. Responses to CM vaccination were also maintained at higher magnitudes over time (13 weeks) compared to other vaccination regimens. Thus, while a ChAdOx1 prime combined with MVA booster vaccination generated stronger and more sustained T-cell responses compared to three DNA vaccinations, the ChAdOx1 primed responses were more narrowly targeted. In conclusion, our findings suggest that the choice of vaccine vectors and prime-boost regimens plays a crucial role in determining the strength, duration, breadth, and focus of T-cell responses, providing further guidance for selecting vaccination strategies.
In vitro antibacterial activity of antiretroviral drugs on key commensal bacteria from the human microbiota
Authors: Elisa Rubio-Garcia, Núria Ferrando, Núria Martin, Clara Ballesté-Delpierre, Jose M. Miró, Roger Paredes, Climnet Casals-Pascual, Jordi Vila
ABSTRACT:
Introduction
Antiretroviral therapy has improved life expectancy in HIV-infected patients. However, people living with HIV under antiretroviral therapy are at higher risks of developing chronic complications and acquiring multidrug resistant bacteria than healthy population. These factors have been associated with shifts in gut microbiome composition and immune activation. It is unclear how antiretroviral drugs affect gut microbiota composition, but it has been observed that antiretroviral treatment is not able to fully restore gut health after HIV infection. Additionally, some antiretroviral drugs have shown antibacterial activity suggesting that these drugs could have a direct impact on the human microbiome composition.
Methods
We determined the in vitro antibacterial activity of 16 antiretroviral drugs against a set of key clinically relevant and human commensal bacterial strains.
Results
Our results demonstrate that 5 antiretroviral drugs have in vitro antibacterial activity against gut and vaginal human commensal bacteria. Zidovudine has antibacterial activity against Escherichia coli, Klebsiella pneumoniae and Prevotella bivia, abacavir against Gardnerella vaginalis, efavirenz against G. vaginalis and P. bivia and bictegravir against Enterococcus spp. and G. vaginalis. Moreover, we describe for the first time that elvitegravir has antibacterial activity against G. vaginalis and P. bivia and, most importantly, against vancomycin-resistant Enterococcus spp. and methicillin-resistant Staphylococcus aureus strains with MIC values of 4-16 and 4 µg/mL, respectively showing high level of effectiveness against the tested multidrug-resistant bacteria.
Discussion
Our results underscore that some antiretroviral drugs may influence the human microbiota composition. In addition, we report the potential use of elvitegravir to treat multidrug-resistant Gram-positive bacteria warranting the need of clinical studies to repurpose this antiretroviral drug.
DOI: https://doi.org/10.3389/fcimb.2023.1306430
JOURNAL: Frontiers in Cellular and Infection Microbiology
DATE: 2024 Jan 08
Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels
Authors:Aleix Elizalde-Torrent, Alessandra Borgognone,Maria Casadellà ,Luis Romero-Martin,Tuixent Escribà,Mariona Parera, Yaiza Rosales-Salgado, Jorge Díaz-Pedroza, Francesc Català-Moll, Marc Noguera-Julian, Christian Brander, Roger Paredes and Alex Olvera.
Abstract: Despite the important role of gut microbiota in the maturation of the immune system, little is known about its impact on the development of T-cell responses to vaccination. Here, we immunized C57BL/6 mice with a prime-boost regimen using DNA plasmid, the Chimpanzee Adenovirus, and the modified Vaccinia Ankara virus expressing a candidate HIV T-cell immunogen and compared the T-cell responses between individuals with an intact or antibiotic-depleted microbiota. Overall, the depletion of the gut microbiota did not result in significant differences in the magnitude or breadth of the immunogen-specific IFNγ T-cell response after vaccination. However, we observed marked changes in the serum levels of four cytokines after vaccinating microbiota-depleted animals, particularly a significant reduction in IL-22 levels. Interestingly, the level of IL-22 in serum correlated with the abundance of Roseburia in the large intestine of mice in the mock and vaccinated groups with intact microbiota. This short-chain fatty acid (SCFA)-producing bacterium was significantly reduced in the vaccinated, microbiota-depleted group. Therefore, our results indicate that, although microbiota depletion reduces serum levels of IL-22, the powerful vaccine regime used could have overcome the impact of microbiota depletion on IFNγ-producing T-cell responses.
Vaccination with an HIV T- cell immunogen induces alterations in the mouse gut microbiota
Authors: Alessandra Borgognone; Aleix Elizalde- Torrent; Maria Casadellà; Luis Romero; Tuixent Escribà; Mariona Parera; Francesc Català-Moll; Marc Noguera- Julian; Christian Brander; Alex Olvera; Roger Paredes
ABSTRACT:
The gut microbiota is emerging as a crucial factor modulating vaccine responses; however, few studies have investigated if vaccines, in turn, can alter the microbiota and to what extent such changes may improve vaccine efficacy. To understand the effect of T-cell vaccination on the gut microbiome, we administered an HIV-1 T-cell immunogen (HTI arm) or PBS (control, Mock arm) to C57Bl/6 mice following a heterologous prime-boost scheme. The longitudinal dynamics of the mice gut microbiota was characterized by 16 S ribosomal RNA sequencing in fecal samples collected from cages, as well as from three gut sections (cecum, small and large intestine). Serum and spleen cells were obtained at the last time point of the study to assess immune correlates using IFNγ ELISPOT and cytokine Luminex® assays. Compared with Mock, HTI-vaccinated mice were enriched in Clostridiales genera (Eubacterium xylanophilum group, Roseburia and Ruminococcus) known as primary contributors of anti-inflammatory metabolites, such as short-chain fatty acids. Such shift was observed after the first HTI dose and remained throughout the study follow-up (18 weeks). However, the enriched Clostridiales genera were different between feces and gut sections. The abundance of bacteria enriched in vaccinated animals positively correlated with HTI-specific T-cell responses and a set of pro-inflammatory cytokines, such as IL-6. This longitudinal analysis indicates that, in mice, T-cell vaccination may promote an increase in gut bacteria known to produce anti-inflammatory molecules, which in turn correlate with proinflammatory cytokines, suggesting an adaptation of the gut microbial milieu to T-cell-induced systemic inflammation.
DOI: https://doi.org/10.1038/s41522-022-00368-y
JOURNAL: NPJ Biofilms and Microbiomes
DATE: 2022 Dec 30
Probiotic effects on immunity and microbiome in HIV-1 discordant patients
Authors: Carlos Blázquez- Bondia; Mariona Parera; Francesc Català-Moll; Maria Casadellà; Aleix Elizalde- Torrent; Meritxell Aguiló; Jordi Espadaler- Mazo; José Ramon Santos; Roger Paredes; Marc Noguera-Julian
ABSTRACT:
Background: Some HIV-1 infected patients are unable to completely recover normal CD4+ T-cell (CD4+) counts after achieving HIV-1 suppression with combined Antiretroviral Therapy (cART), hence being classified as immuno-discordant. The human microbiome plays a crucial role in maintaining immune homeostasis and is a potential target towards immune reconstitution.
Setting: RECOVER (NCT03542786) was a double-blind placebo-controlled clinical trial designed to evaluate if the novel probiotic i3.1 (AB-Biotics, Sant Cugat del Vallès, Spain) was able to improve immune reconstitution in HIV-1 infected immuno-discordant patients with stable cART and CD4+ counts <500 cells/mm3. The mixture consisted of two strains of L. plantarum and one of P. acidilactici, given with or without a fiber-based prebiotic.
Methods: 71 patients were randomized 1:2:2 to Placebo, Probiotic or probiotic + prebiotic (Synbiotic), and were followed over 6 months + 3-month washout period, in which changes on systemic immune status and gut microbiome were evaluated. Primary endpoints were safety and tolerability of the investigational product. Secondary endpoints were changes on CD4+ and CD8+ T-cell (CD8+) counts, inflammation markers and faecal microbiome structure, defined by alpha diversity (Gene Richness), beta diversity (Bray-Curtis) and functional profile. Comparisons across/within groups were performed using standard/paired Wilcoxon test, respectively.
Results: Adverse event (AE) incidence was similar among groups (53%, 33%, and 55% in the Placebo, Probiotic and Synbiotic groups, respectively, the most common being grade 1 digestive AEs: flatulence, bloating and diarrhoea. Two grade 3 AEs were reported, all in the Synbiotic group: abdominal distension (possibly related) and malignant lung neoplasm (unrelated), and 1 grade 4 AE in the Placebo: hepatocarcinoma (unrelated). Synbiotic exposure was associated with a higher increase in CD4+/CD8+ T-cell (CD4/CD8) ratio at 6 months vs baseline (median=0.76(IQR=0.51) vs 0.72(0. 45), median change= 0.04(IQR=0.19), p = 0.03). At month 9, the Synbiotic group had a significant increase in CD4/CD8 ratio (0.827(0.55) vs 0.825(0.53), median change = 0.04(IQR=0.15), p= 0.02) relative to baseline, and higher CD4+ counts (447 (157) vs. 342(73) counts/ml, p = 0.03), and lower sCD14 values (2.16(0.67) vs 3.18(0.8), p = 0.008) than Placebo. No effect in immune parameters was observed in the Probiotic arm. None of the two interventions modified microbial gene richness (alpha diversity). However, intervention as categorical variable was associated with slight but significant effect on Bray-Curtis distance variance (Adonis R2 = 0.02, p = 0.005). Additionally, at month 6, Synbiotic intervention was associated with lower pathway abundances vs Placebo of Assimilatory Sulphate Reduction (8.79·10-6 (1.25·10-5) vs. 1.61·10-5 (2.77·10-5), p = 0.03) and biosynthesis of methionine (2.3·10-5 (3.17·10-5) vs. 4·10-5 (5.66·10-5), p = 0.03) and cysteine (1.83·10-5 (2.56·10-5) vs. 3.3·10-5 (4.62·10-5), p = 0.03). At month 6, probiotic detection in faeces was associated with significant decreases in C Reactive Protein (CRP) vs baseline (11.1(22) vs. 19.2(66), median change= -2.7 (13.2) ug/ml, p = 0.04) and lower IL-6 values (0.58(1.13) vs. 1.17(1.59) ug/ml, p = 0.02) when compared with samples with no detectable probiotic. No detection of the probiotic was associated with higher CD4/CD8 ratio at month 6 vs baseline (0.718(0.57) vs. 0.58(0.4), median change = 0.4(0.2), p = 0.02). After washout, probiotic non-detection was also associated with a significant increase in CD4+ counts (457(153) vs. 416(142), median change = 45(75), counts/ml, p = 0.005) and CD4/CD8 ratio (0.67(0.5) vs 0.59(0.49), median change = 0.04 (0.18), p = 0.02).
Conclusion: A synbiotic intervention with L. plantarum and P. acidilactici was safe and led to small increases in CD4/CD8 ratio and minor reductions in sCD14 of uncertain clinical significance. A probiotic with the same composition was also safe but did not achieve any impact on immune parameters or faecal microbiome composition.
Safety, immunogenicity and effect on viral rebound of HTI vaccines in early treated HIV-1 infection: a randomized, placebo-controlled phase 1 trial
Authors: Lucia Bailón, Anuska Llano, Samandhy Cedeño, Tuixent Escribà, Miriam Rosás-Umbert, Mariona Parera, Maria Casadellà, Miriam Lopez, Francisco Pérez, Bruna Oriol-Tordera, Marta Ruiz-Riol, Josep Coll, Felix Perez, Àngel Rivero, Anne R. Leselbaum, Ian McGowan, Devi Sengupta, Edmund G. Wee, Tomáš Hanke, Roger Paredes, Yovaninna Alarcón-Soto, Bonaventura Clotet, Marc Noguera-Julian, Christian Brander, the AELIX002 Study Group
Abstract:
HIVACAT T-cell immunogen (HTI) is a novel human immunodeficiency virus (HIV) vaccine immunogen designed to elicit cellular immune responses to HIV targets associated with viral control in humans. The AELIX-002 trial was a randomized, placebo-controlled trial to evaluate as a primary objective the safety of a combination of DNA.HTI (D), MVA.HTI (M) and ChAdOx1.HTI (C) vaccines in 45 early-antiretroviral (ART)-treated individuals (44 men, 1 woman; NCT03204617). Secondary objectives included T-cell immunogenicity, the effect on viral rebound and the safety of an antiretroviral treatment interruption (ATI). Adverse events were mostly mild and transient. No related serious adverse events were observed. We show here that HTI vaccines were able to induce strong, polyfunctional and broad CD4 and CD8 T-cell responses. All participants experienced detectable viral rebound during ATI, and resumed ART when plasma HIV-1 viral load reached either >100,000 copies ml−1, >10,000 copies ml−1 for eight consecutive weeks, or after 24 weeks of ATI. In post-hoc analyses, HTI vaccines were associated with a prolonged time off ART in vaccinees without beneficial HLA (human leukocyte antigen) class I alleles. Plasma viral load at the end of ATI and time off ART positively correlated with vaccine-induced HTI-specific T-cell responses at ART cessation. Despite limited efficacy of the vaccines in preventing viral rebound, their ability to elicit robust T-cell responses towards HTI may be beneficial in combination cure strategies, which are currently being tested in clinical trials.
Gut microbiome signatures linked to HIV-1 reservoir size and viremia control
Authors: Alessandra Borgognone; Bruna Oriol- Tordera; Beatriz Mothe; Roger Paredes; Maria C. Puertas
ABSTRACT:
Background
The potential role of the gut microbiome as a predictor of immune-mediated HIV-1 control in the absence of antiretroviral therapy (ART) is still unknown. In the BCN02 clinical trial, which combined the MVA.HIVconsv immunogen with the latency-reversing agent romidepsin in early-ART treated HIV-1 infected individuals, 23% (3/13) of participants showed sustained low-levels of plasma viremia during 32 weeks of a monitored ART pause (MAP). Here, we present a multi-omics analysis to identify compositional and functional gut microbiome patterns associated with HIV-1 control in the BCN02 trial.
Results
Viremic controllers during the MAP (controllers) exhibited higher Bacteroidales/Clostridiales ratio and lower microbial gene richness before vaccination and throughout the study intervention when compared to non-controllers. Longitudinal assessment indicated that the gut microbiome of controllers was enriched in pro-inflammatory bacteria and depleted in butyrate-producing bacteria and methanogenic archaea. Functional profiling also showed that metabolic pathways related to fatty acid and lipid biosynthesis were significantly increased in controllers. Fecal metaproteome analyses confirmed that baseline functional differences were mainly driven by Clostridiales. Participants with high baseline Bacteroidales/Clostridiales ratio had increased pre-existing immune activation-related transcripts. The Bacteroidales/Clostridiales ratio as well as host immune-activation signatures inversely correlated with HIV-1 reservoir size.
Conclusions
The present proof-of-concept study suggests the Bacteroidales/Clostridiales ratio as a novel gut microbiome signature associated with HIV-1 reservoir size and immune-mediated viral control after ART interruption.
Epigenetic landscape in the kick-and-kill therapeutic vaccine BCN02 clinical trial is associated with antiretroviral treatment interruption (ATI) outcome
Authors: Bruna Oriol-Tordera, Anna Esteve-Codina, María Berdasco, Míriam Rosas-Umbert, Elena Goncalves, Clara Duran-Castells, Francesc Catala-Moll, Anuska Llano, Samandhy Cedeno, Maria C. Puertas, Martin Tolstrup, Ole S. Søgaard, Bonaventura Clotet, Javier Martínez-Picado, Tomas Hanke, Behazine Combadiere, Roger Paredes, Dennis Hartigan-O’Connor, Manel Esteller, Michael Meulbroek, María Luz Calle, Alex Sanchez-Pla, Jose Molto, Beatriz Mothe, Christian Brander, and Marta Ruiz-Riol
ABSTRACT:
Background
The BCN02-trial combined therapeutic vaccination with a viral latency reversing agent (romidepsin, RMD) in HIV-1-infected individuals and included a monitored antiretroviral pause (MAP) as an efficacy read-out identifying individuals with an early or late (< or > 4weeks) viral-rebound. Integrated -omics analyses were applied prior treatment interruption to identify markers of virus control during MAP.
Methods
PBMC, whole-genome DNA methylation and transcriptomics were assessed in 14 BCN02 participants, including 8 Early and 4 Late viral-rebound individuals. Chromatin state, histone marks and integration analysis (histone-3 acetylation (H3Ac), viral load, proviral levels and HIV-specific T cells responses) were included. REDUC-trial samples (n = 5) were included as a control group for RMD administration alone.
Findings
DNA methylation imprints after receiving the complete intervention discriminated Early versus Late viral-rebound individuals before MAP. Also, differential chromatin accessibility and histone marks at DNA methylation level were detected. Importantly, the differential DNA methylation positions (DMPs) between Early and Late rebounders before MAP were strongly associated with viral load, proviral levels as well as the HIV-specific T-cell responses. Most of these DMPs were already present prior to the intervention and accentuated after RMD infusion.
Interpretation
This study identifies host DNA methylation profiles and epigenetic cascades that are predictive of subsequent virus control in a kick-and-kill HIV cure strategy.
Host Transcriptome and Microbiota Signatures Prior to Immunization Profile Vaccine Humoral Responsiveness
Authors: Elena Golçalves, Yolanda Guillén, Javier R.Lama, Jorge Sanchez, Christian Brander, Roger Paredes, Behazine Combadière
Abstract
The identification of new biomarkers is essential to predict responsiveness to vaccines. We investigated the whole-blood transcriptome and microbiome prior to immunization, in order to assess their involvement in induction of humoral responses two months later. We based our analyses on stool and skin microbiota, and blood transcriptome prior to immunization, in a randomized clinical study in which participants were vaccinated with the MVA-HIV clade B vaccine (MVA-B). We found that the levels of neutralizing antibody responses were correlated with abundance of Eubacterium in stool and Prevotella in skin. In addition, genus diversity and bacterial species abundance were also correlated with the expression of genes involved in B cell development prior to immunization and forecast strong responders to MVA-B. To our knowledge, this is the first study integrating host blood gene expression and microbiota that might open an avenue of research in this field and to optimize vaccination strategies and predict responsiveness to vaccines.
PREVIOUS RELATED PUBLICATIONS
Associations of the gut microbiome and clinical factors with acute GVHD in allogeneic HSCT recipients
AUTHORS: Ilett EE, Jørgensen M, Noguera-Julian M, Nørgaard JC, Daugaard G, Helleberg M, Paredes R, Murray DD, Lundgren J, MacPherson C, Reekie J, Sengeløv H.
ABSTRACT:
Acute graft-versus-host disease (aGVHD) is a leading cause of transplantation-related mortality after allogeneic hematopoietic stem cell transplantation (aHSCT). 16S ribosomal RNA (16S rRNA) gene-based studies have reported that lower gut bacterial diversity and the relative abundance of certain bacteria after aHSCT are associated with aGVHD. Using shotgun metagenomic sequencing and a large cohort, we aimed to confirm and extend these observations. Adult aHSCT recipients with stool samples collected from day -30 to day 100 relative to aHSCT were included. One sample was selected per patient per period (pre-aHSCT (day -30 to day 0), early post-aHSCT (day 1 to day 28), and late post-aHSCT (day 29 to day 100)), resulting in 150 aHSCT recipients and 259 samples. Microbial and clinical factors were tested for differences between time periods and an association with subsequent aGVHD. Patients showed a decline in gut bacterial diversity posttransplant, with several patients developing a dominance of Enterococcus. A total of 36 recipients developed aGVHD at a median of 34 days (interquartile range, 26-50 days) post-aHSCT. Lower microbial gene richness (P = .02), a lower abundance of the genus Blautia (P = .05), and a lower abundance of Akkermansia muciniphila (P = .01) early post-aHSCT was observed in those who developed aGVHD. Myeloablative conditioning was associated with aGVHD along with a reduction in gene richness and abundance of Blautia and A muciniphila. These results confirm low diversity and Blautia being associated with aGVHD. Crucially, we add that pretransplant conditioning is associated with changes in gut microbiota. Investigations are warranted to determine the interplay of gut microbiota and conditioning in the development of aGVHD.
The Impact of Human Immunodeficiency Virus Infection on Gut Microbiota α-Diversity: An Individual-level Meta-analysis
AUTHORS: Tuddenham SA, Koay WLA, Zhao N, White JR, Ghanem KG, Sears CL; HIV Microbiome Re-analysis Consortium
ABSTRACT:
Background: Whether human immunodeficiency virus (HIV) infection impacts gut microbial α-diversity is controversial. We reanalyzed raw 16S ribosomal RNA (rRNA) gene sequences and metadata from published studies to examine α-diversity measures between HIV-uninfected (HIV-) and HIV-infected (HIV+) individuals.
Methods: We conducted a systematic review and individual level meta-analysis by searching Embase, Medline, and Scopus for original research studies (inception to 31 December 2017). Included studies reported 16S rRNA gene sequences of fecal samples from HIV+ patients. Raw sequence reads and metadata were obtained from public databases or from study authors. Raw reads were processed through standardized pipelines with use of a high-resolution taxonomic classifier. The χ2 test, paired t tests, and generalized linear mixed models were used to relate α-diversity measures and clinical metadata.
Results: Twenty-two studies were identified with 17 datasets available for analysis, yielding 1032 samples (311 HIV-, 721 HIV+). HIV status was associated with a decrease in measures of α-diversity (P < .001). However, in stratified analysis, HIV status was associated with decreased α-diversity only in women and in men who have sex with women (MSW) but not in men who have sex with men (MSM). In analyses limited to women and MSW, controlling for HIV status, women displayed increased α-diversity compared with MSW.
Conclusions: Our study suggests that HIV status, sexual risk category, and gender impact gut microbial community α-diversity. Future studies should consider MSM status in gut microbiome analyses.
Gut microbiome comparability of fresh-frozen versus stabilized-frozen samples from hospitalized patients using 16S rRNA gene and shotgun metagenomic sequencing
AUTHORS: Ilett EE, Jørgensen M, Noguera-Julian M, Daugaard G, Murray DD, Helleberg M, Paredes R, Lundgren J, Sengeløv H, MacPherson C.
ABSTRACT:
Collection of faecal samples for microbiome analysis in acutely sick patients is logistically difficult, particularly if immediate freezing is required (i.e. fresh-frozen, or FF sampling). Previous studies in healthy/non-hospitalized volunteers have shown that chemical stabilization (i.e. stabilized-frozen, or SF sampling) allows room-temperature storage with comparable results to FF samples. To test this in a hospital setting we compared FF and SF approaches across 17 patients undergoing haematopoietic stem cell transplantation (HSCT) using both 16S rRNA gene and shotgun metagenomic sequencing. A paired (same stool specimen) comparison of FF and SF samples was made, with an overall comparable level in relative taxonomic abundances between the two sampling techniques. Though shotgun metagenomic sequencing found significant differences for certain bacterial genera (P < 0.001), these were considered minor methodological effects. Within-sample diversity of either method was not significantly different (Shannon diversity P16SrRNA = 0.68 and Pshotgun = 0.89) and we could not reject the null hypothesis that between-sample variation in FF and SF were equivalent (P16SrRNA = 0.98 and Pshotgun = 1.0). This indicates that SF samples can be used to reliably study the microbiome in acutely sick patient populations, thus creating and enabling further outcomes-based metagenomic studies on similarly valuable cohorts.
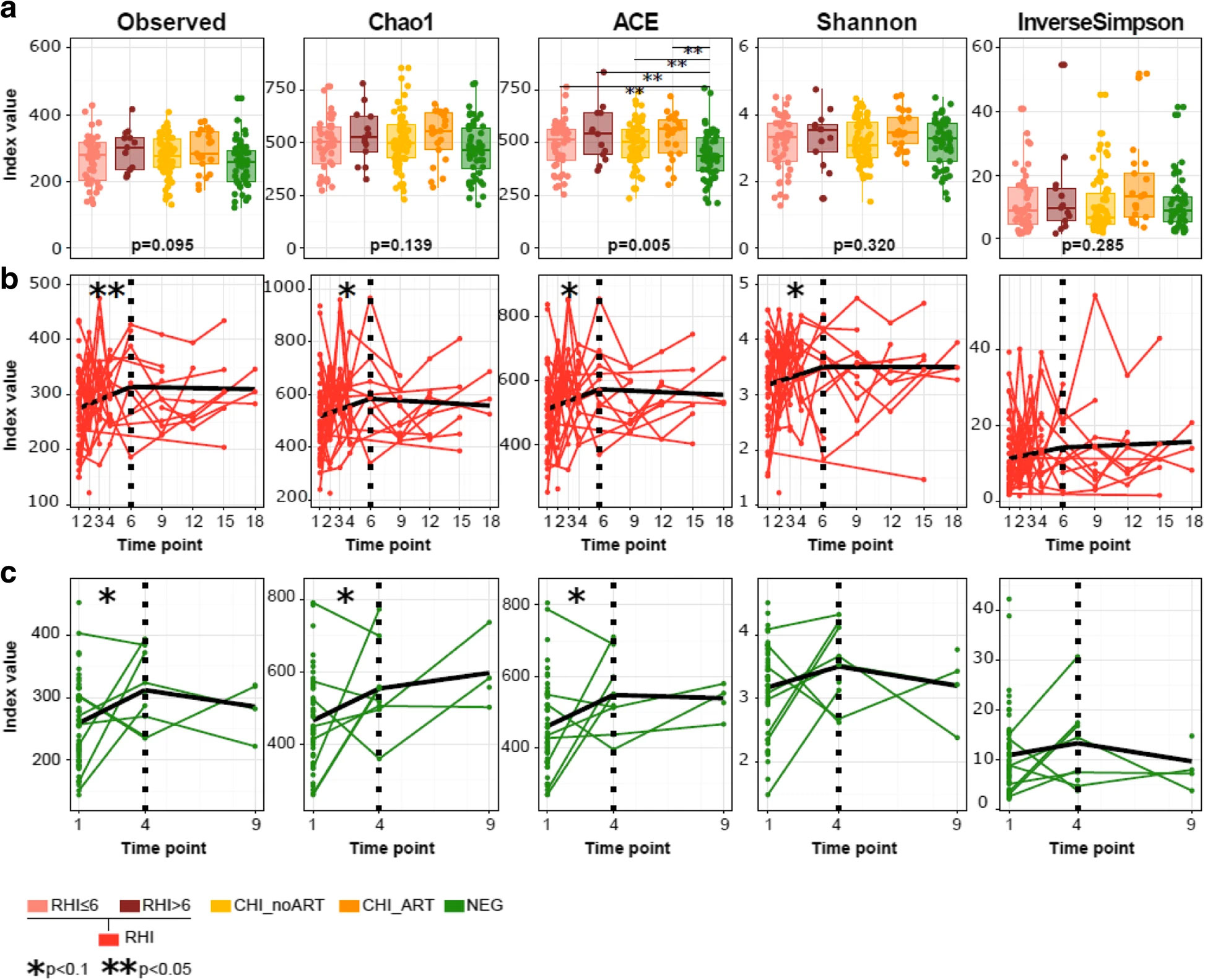
Evolution of the gut microbiome following acute HIV-1 infection
AUTHORS: Rocafort M, Noguera-Julian M, Rivera J, Pastor L, Guillén Y, Langhorst J, Parera M, Mandomando I, Carrillo J, Urrea V, Rodríguez C, Casadellà M, Calle ML, Clotet B, Blanco J, Naniche D, Paredes R.
ABSTRACT:
Background: In rhesus macaques, simian immunodeficiency virus infection is followed by expansion of enteric viruses but has a limited impact on the gut bacteriome. To understand the longitudinal effects of HIV-1 infection on the human gut microbiota, we prospectively followed 49 Mozambican subjects diagnosed with recent HIV-1 infection (RHI) and 54 HIV-1-negative controls for 9-18 months and compared them with 98 chronically HIV-1-infected subjects treated with antiretrovirals (n = 27) or not (n = 71).
Results: We show that RHI is followed by increased fecal adenovirus shedding, which persists during chronic HIV-1 infection and does not resolve with ART. Recent HIV-1 infection is also followed by transient non-HIV-specific changes in the gut bacterial richness and composition. Despite early resilience to change, an HIV-1-specific signature in the gut bacteriome-featuring depletion of Akkermansia, Anaerovibrio, Bifidobacterium, and Clostridium-previously associated with chronic inflammation, CD8+ T cell anergy, and metabolic disorders, can be eventually identified in chronically HIV-1-infected subjects.
Conclusions: Recent HIV-1 infection is associated with increased fecal shedding of eukaryotic viruses, transient loss of bacterial taxonomic richness, and long-term reductions in microbial gene richness. An HIV-1-associated microbiome signature only becomes evident in chronically HIV-1-infected subjects.
Low nadir CD4+ T-cell counts predict gut dysbiosis in HIV-1 infection
AUTHORS: Guillén Y, Noguera-Julian M, Rivera J, Casadellà M, Zevin AS, Rocafort M, Parera M, Rodríguez C, Arumí M, Carrillo J, Mothe B, Estany C, Coll J, Bravo I, Herrero C, Saz J, Sirera G, Torrella A, Navarro J, Crespo M, Negredo E, Brander C, Blanco J, Calle ML, Klatt NR, Clotet B, Paredes R.
ABSTRACT:
Human immunodeficiency virus (HIV)-1 infection causes severe gut and systemic immune damage, but its effects on the gut microbiome remain unclear. Previous shotgun metagenomic studies in HIV-negative subjects linked low-microbial gene counts (LGC) to gut dysbiosis in diseases featuring intestinal inflammation. Using a similar approach in 156 subjects with different HIV-1 phenotypes, we found a strong, independent, dose-effect association between nadir CD4+ T-cell counts and LGC. As in other diseases involving intestinal inflammation, the gut microbiomes of subjects with LGC were enriched in gram-negative Bacteroides, acetogenic bacteria and Proteobacteria, which are able to metabolize reactive oxygen and nitrogen species; and were depleted in oxygen-sensitive methanogenic archaea and sulfate-reducing bacteria. Interestingly, subjects with LGC also showed increased butyrate levels in direct fecal measurements, consistent with enrichment in Roseburia intestinalis despite reductions in other butyrate producers. The microbiomes of subjects with LGC were also enriched in bacterial virulence factors, as well as in genes associated with beta-lactam, lincosamide, tetracycline, and macrolide resistance. Thus, low nadir CD4+ T-cell counts, rather than HIV-1 serostatus per se, predict the presence of gut dysbiosis in HIV-1 infected subjects. Such dysbiosis does not display obvious HIV-specific features; instead, it shares many similarities with other diseases featuring gut inflammation.
Balances: a New Perspective for Microbiome Analysis
AUTHORS: Rivera-Pinto J, Egozcue JJ, Pawlowsky-Glahn V, Paredes R, Noguera-Julian M, Calle ML.
ABSTRACT:
High-throughput sequencing technologies have revolutionized microbiome research by allowing the relative quantification of microbiome composition and function in different environments. In this work we focus on the identification of microbial signatures, groups of microbial taxa that are predictive of a phenotype of interest. We do this by acknowledging the compositional nature of the microbiome and the fact that it carries relative information. Thus, instead of defining a microbial signature as a linear combination in real space corresponding to the abundances of a group of taxa, we consider microbial signatures given by the geometric means of data from two groups of taxa whose relative abundances, or balance, are associated with the response variable of interest. In this work we present selbal, a greedy stepwise algorithm for selection of balances or microbial signatures that preserves the principles of compositional data analysis. We illustrate the algorithm with 16S rRNA abundance data from a Crohn’s microbiome study and an HIV microbiome study. IMPORTANCE We propose a new algorithm for the identification of microbial signatures. These microbial signatures can be used for diagnosis, prognosis, or prediction of therapeutic response based on an individual’s specific microbiota.
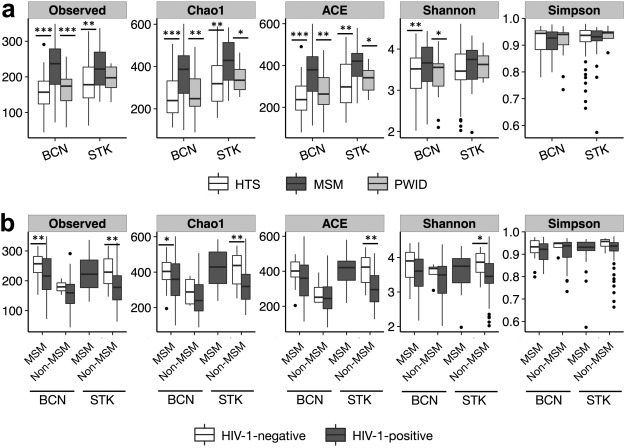
Gut Microbiota Linked to Sexual Preference and HIV Infection
AUTHORS: Noguera-Julian M, Rocafort M, Guillén Y, Rivera J, Casadellà M, Nowak P, Hildebrand F, Zeller G, Parera M, Bellido R, Rodríguez C, Carrillo J, Mothe B, Coll J, Bravo I, Estany C, Herrero C, Saz J, Sirera G, Torrela A, Navarro J, Crespo M, Brander C, Negredo E, Blanco J, Guarner F, Calle ML, Bork P, Sönnerborg A, Clotet B, Paredes R.
ABSTRACT:
The precise effects of HIV-1 on the gut microbiome are unclear. Initial cross-sectional studies provided contradictory associations between microbial richness and HIV serostatus and suggested shifts from Bacteroides to Prevotella predominance following HIV-1 infection, which have not been found in animal models or in studies matched for HIV-1 transmission groups. In two independent cohorts of HIV-1-infected subjects and HIV-1-negative controls in Barcelona (n = 156) and Stockholm (n = 84), men who have sex with men (MSM) predominantly belonged to the Prevotella-rich enterotype whereas most non-MSM subjects were enriched in Bacteroides, independently of HIV-1 status, and with only a limited contribution of diet effects. Moreover, MSM had a significantly richer and more diverse fecal microbiota than non-MSM individuals. After stratifying for sexual orientation, there was no solid evidence of an HIV-specific dysbiosis. However, HIV-1 infection remained consistently associated with reduced bacterial richness, the lowest bacterial richness being observed in subjects with a virological-immune discordant response to antiretroviral therapy. Our findings indicate that HIV gut microbiome studies must control for HIV risk factors and suggest interventions on gut bacterial richness as possible novel avenues to improve HIV-1-associated immune dysfunction.
RESOURCES
